What should your doctor know?
You can expect that your doctor understands the Center for Disease Control's training on Hemochromatosis. The field of Genomics - in which the gene related specifics of iron regulation are intrinsically tied - moves at an alarming rate. Doctors should not be expected to understand or even have heard of the latest research on Genetic Hemochromatosis. Doctors are specialists in biochemistry which is a very different discipline than genetics. They should at least understand the CDC training AND be willing to listen to you about what you have discovered on the internet. Yes, even if it is from Wikipedia! If you suspect they are not following this training, then you should not feel too intimidated to mention it to them. The training is at: CDC Hemochromatosis Training.
The summary of what doctors are looking for according to this training is:
Testing Protocol
The summary of what doctors are looking for according to this training is:
- Recommended laboratory iron tests for the workup of a patient you suspect may have hemochromatosis are:
- Fasting transferrin saturation test (TS).
- Serum ferritin test (SF).
Testing Protocol
- Transferrin saturation (TS).
- Fasting values >45% should be followed by a serum ferritin test and additional workup.
- Serum ferritin (SF).
- Values >200 ng/mL for premenopausal females OR >300 ng/mL for postmenopausal females and males indicate iron overload; phlebotomy treatment is warranted in the absence of other causes.
- SF values can be elevated with liver disease, inflammation, and neoplasm.
What do these tests mean?
TS can be compared to an old amusement park ride. It measures the amount of iron riding around in your blood. Just like a roller coaster, you do not want to overload the cars with passengers beyond the capacity of the ride. In this case if our body allows more than 45% of the cars to be filled with passengers then some damage can start to occur like free radicals. The body should not allow that to happen, but if it does then Genetic Hemochromatosis could be the cause. When I was diagnosed my TS was 96.
SF is basically an indicator of how much iron is stored in the body. Under 50 ng/mL could indicate anemia while over 150 ng/mL is elevated - meaning that is when it can indicate liver disease, inflammation, and neoplasm. This is different then the numbers listed above because here we are discussing the optimal value - not the value which indicates overload. Values over 200 ng/mL are severely elevated even if they are not high enough to be diagnosed as Iron Overload. Remember that Iron Overload is synonymous with Hemochromatosis, but not necessarily Genetic Hemochromatosis. When I was diagnosed my SF was 296. I had been donating blood for years prior to diagnosis which is likely the reason this number was so low when I was 39. My Serum Iron was 271. This is not the same thing. Serum Iron is all about iron in the blood while serum ferritin is about iron in the body.
If both these values are elevated, the doctor should order a genetic test to test for Hereditary Hemochromatosis.
SF is basically an indicator of how much iron is stored in the body. Under 50 ng/mL could indicate anemia while over 150 ng/mL is elevated - meaning that is when it can indicate liver disease, inflammation, and neoplasm. This is different then the numbers listed above because here we are discussing the optimal value - not the value which indicates overload. Values over 200 ng/mL are severely elevated even if they are not high enough to be diagnosed as Iron Overload. Remember that Iron Overload is synonymous with Hemochromatosis, but not necessarily Genetic Hemochromatosis. When I was diagnosed my SF was 296. I had been donating blood for years prior to diagnosis which is likely the reason this number was so low when I was 39. My Serum Iron was 271. This is not the same thing. Serum Iron is all about iron in the blood while serum ferritin is about iron in the body.
If both these values are elevated, the doctor should order a genetic test to test for Hereditary Hemochromatosis.
The Genetic Test
In the United States, most doctors order a genetic test that looks for 2 SNPs - C282Y and H64D. We can have two copies of either or none at all. The test returns Positive if there are 2 copies of C282Y or a copy of C282Y and one of H64D. Do not just get a positive or negative response from the doctor. You will want a copy of these results regardless of what they are. It is very important to realize that new research is proving that even if this test comes back Negative, you may still have Hereditary Hemochromatosis. So remember - Positive is is always Positive, but Negative can be Positive too! Much more on this can be found on my Genetics Terms page.
What does the Genetic Test really mean?
I describe the genes in detail in my Genetics Terms page. Here is a good analogy to help you and your doctor understand. Lets call iron overload a video game. In this game iron moves from left to right on the computer screen and tries to load onto the right side. The percentage of iron loaded onto the right side is your TS score. What is really cool is that you get to use two mice to click on the iron as it crosses the screen. The goal is to have the lowest TS score when time expires. Now comes the catch, our DNA is just a blueprint for building a protein. In this analogy the protein is the computer mouse. A HFE gene with the C282Y blueprint will create a mouse with a button that is sealed inside the mouse making it totally unclickable. The H63D (and to some extent the S65C) blueprint will have the button in the right place, but they make the surface really slippery. So if you have two C282Y mice ... then you just watch the iron march across the screen and load the TS score up into the 90s. If you have one C282Y mouse and one H63D mouse, then you can stop some of the iron but it does not work very well at all. You can imagine the different combinations and get an idea of what happens.
Now here is the trick. No one can see the mouse. We know how the mouse acts and think we know what it looks like, but all anyone can see is the TS score and the blueprint. So if the TS score is high and the blueprint does not show a C282Y, H63D or S65C mutation we have two choices, we can either assume the mouse is perfect and something else is wrong with the computer or we can infer that there is something else wrong with the blueprint for the mouse that we cannot see and try to identify what that is.
Here are the details behind this analogy - the HFE protein must express itself through the cell wall, but with the C282Y mutation a sulfide bond is missing and it is not strong enough to get through (so the button does not show on the outside of the mouse). With a H63D mutation, a polar charged molecule is replaced by a polar uncharged molecule on the binding location with the TFR2 protein (making the bond between the proteins different like having a slippery mouse button). The S65C mutation is only two amino acids away from H63D, so this is the same area where the TFR2 protein binds. The normal polar charged molecule here is changed out for a special molecule with a sulfur atom at the end. Again this changes the bonds that can be made with the TFR2 protein. After HFE binds to TFR2, they go off to increase the production of the hepcidin hormone which stops iron from being added into the TS 'score'.
Below is an image of this cycle and a more scientific explanation. The key for us is on the left with the HFE 'button' sticking out of the liver (which sort of looks like the computer mouse in my explanation - well, kind of). For c282y homozygotes, there are no HFE 'buttons' sticking out of the liver. For c282y heterozygotes, there are only half as many HFE 'buttons'. For h63d, they are there but are not shaped quite right to bind with Tf and TfR2 so it does not always provide the right signal to HAMP in order to create Hepcidin and stop us from absorbing (and recycling) iron.
Now here is the trick. No one can see the mouse. We know how the mouse acts and think we know what it looks like, but all anyone can see is the TS score and the blueprint. So if the TS score is high and the blueprint does not show a C282Y, H63D or S65C mutation we have two choices, we can either assume the mouse is perfect and something else is wrong with the computer or we can infer that there is something else wrong with the blueprint for the mouse that we cannot see and try to identify what that is.
Here are the details behind this analogy - the HFE protein must express itself through the cell wall, but with the C282Y mutation a sulfide bond is missing and it is not strong enough to get through (so the button does not show on the outside of the mouse). With a H63D mutation, a polar charged molecule is replaced by a polar uncharged molecule on the binding location with the TFR2 protein (making the bond between the proteins different like having a slippery mouse button). The S65C mutation is only two amino acids away from H63D, so this is the same area where the TFR2 protein binds. The normal polar charged molecule here is changed out for a special molecule with a sulfur atom at the end. Again this changes the bonds that can be made with the TFR2 protein. After HFE binds to TFR2, they go off to increase the production of the hepcidin hormone which stops iron from being added into the TS 'score'.
Below is an image of this cycle and a more scientific explanation. The key for us is on the left with the HFE 'button' sticking out of the liver (which sort of looks like the computer mouse in my explanation - well, kind of). For c282y homozygotes, there are no HFE 'buttons' sticking out of the liver. For c282y heterozygotes, there are only half as many HFE 'buttons'. For h63d, they are there but are not shaped quite right to bind with Tf and TfR2 so it does not always provide the right signal to HAMP in order to create Hepcidin and stop us from absorbing (and recycling) iron.
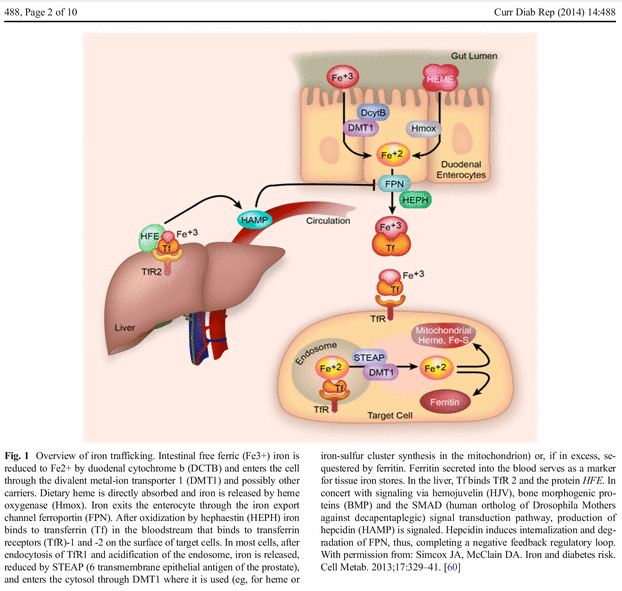
Additionally, a nice description of the whole process can be found here. This is really an article regarding anemia, but everything discussed applies to us as well. And yes we can have anemia as well. In fact, we are essentially kept at a moderate state of iron deficiency anemia when we are in maintenance.
The future testing protocol?
In their paper Novel observations in hereditary hemochromatosis: potential implications for clinical strategies, Dorine W. Swinkels and Robert E. Fleming (the full article is here) recommend the following improvement to the diagnostic protocol:
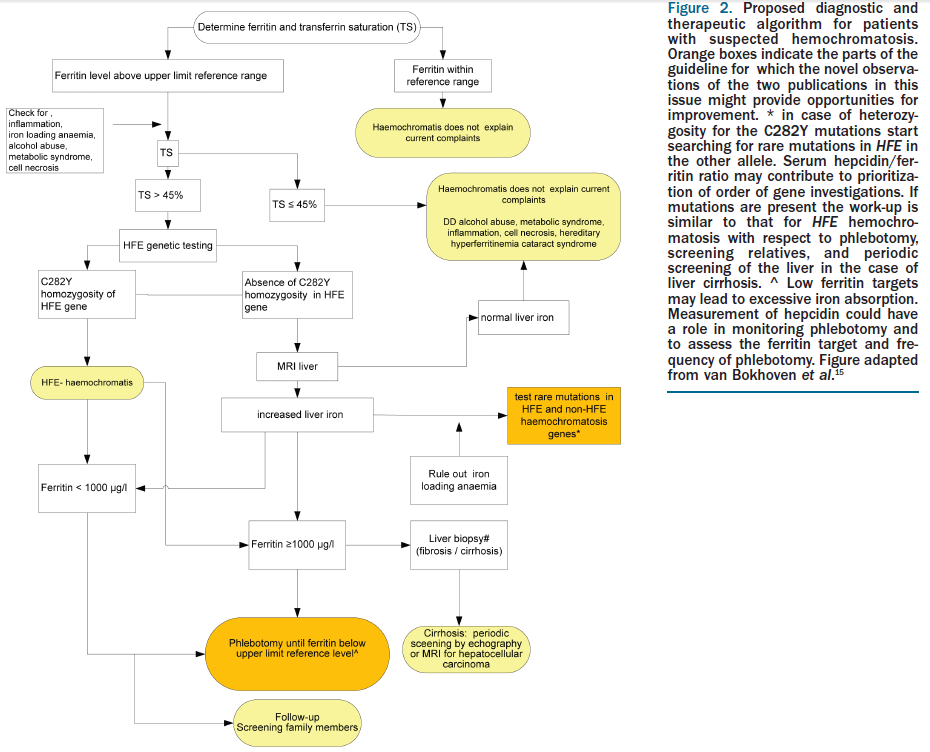
Future definition of Genetic Hemochromatosis?
A Endocrinologist conference in the UK (BES 2013) offered a presentation in March of 2013 with this abstract. I have copied it below and added my emphasis in the event the link breaks after the conference. The authors are Heinz Zoller & Melanie Schranz from the Medical University of Innsbruck, Innsbruck, Austria.
"In contrast to the conventional view of haemochromatosis as a monogenic disease with autosomal recessive inheritance, more recent evidence form genetic, epidemiological, cell biological and clinical studies, challenges this view. The concept that haemochromatosis is an endocrine disorder of mixed etiology embraces the poylgenic nature of the disease, the low penetrance and the similarities in phenotype of genetic and acquired forms of iron overload. Key to understanding haemochromatosis as an endocrine disorder is that iron overload is caused by a deficiency of the iron hormone ‘hepcidin’.
Accordingly, haemochromatosis can be viewed the ‘diabetes of iron metabolism’, where hepcidin and iron have a functional relationship comparable to insulin and glucose. Studies in mice defective for the genes encoding the hemochromatosis protein HFE, transferrin receptor two, and bone morphogenic protein six provide first insight into the ‘iron sensor’, which signals directly to the hepcidin promoter and thus control its transcription rate. Studies on the iron export protein ferroportin and its direct negative regulator hepcidin have shown that plasma iron is regulated via controlled release of iron from recycling macrophages, duodenal enterocytes and hepatocytes. This closes a feedback-loop, where hepcidin tightly controls plasma iron for sufficient delivery to cells utilizing the metal, while avoiding oxidative stress though uncontrolled release of iron into the circulation.
In patients with haemochromatosis, hepcidin production is inappropriately low which causes uncontrolled release of iron into the circulation that results in increased transferrin saturation, which is the principal clinical biochemical defect in hemochromatosis. Genetically hemochromatosis is associated with homozygosity for the common C282Y polymorphism of the HFE gene. Although the exact function of this protein is still unknown, studies in cell and animal models of the disease suggest that HFE is part of the ‘iron sensor’ expressed in hepatocytes. Distribution of iron overload in HFE associated hemochromatosis is similar to other genetic iron overload disorders such as haemochromatosos associated with mutations in the transferrin receptor two gene, hemojuvelin gene or the hepcidin gene, which supports the role of these gene products in the iron sensing mechanism, that is being unravelled."
"In contrast to the conventional view of haemochromatosis as a monogenic disease with autosomal recessive inheritance, more recent evidence form genetic, epidemiological, cell biological and clinical studies, challenges this view. The concept that haemochromatosis is an endocrine disorder of mixed etiology embraces the poylgenic nature of the disease, the low penetrance and the similarities in phenotype of genetic and acquired forms of iron overload. Key to understanding haemochromatosis as an endocrine disorder is that iron overload is caused by a deficiency of the iron hormone ‘hepcidin’.
Accordingly, haemochromatosis can be viewed the ‘diabetes of iron metabolism’, where hepcidin and iron have a functional relationship comparable to insulin and glucose. Studies in mice defective for the genes encoding the hemochromatosis protein HFE, transferrin receptor two, and bone morphogenic protein six provide first insight into the ‘iron sensor’, which signals directly to the hepcidin promoter and thus control its transcription rate. Studies on the iron export protein ferroportin and its direct negative regulator hepcidin have shown that plasma iron is regulated via controlled release of iron from recycling macrophages, duodenal enterocytes and hepatocytes. This closes a feedback-loop, where hepcidin tightly controls plasma iron for sufficient delivery to cells utilizing the metal, while avoiding oxidative stress though uncontrolled release of iron into the circulation.
In patients with haemochromatosis, hepcidin production is inappropriately low which causes uncontrolled release of iron into the circulation that results in increased transferrin saturation, which is the principal clinical biochemical defect in hemochromatosis. Genetically hemochromatosis is associated with homozygosity for the common C282Y polymorphism of the HFE gene. Although the exact function of this protein is still unknown, studies in cell and animal models of the disease suggest that HFE is part of the ‘iron sensor’ expressed in hepatocytes. Distribution of iron overload in HFE associated hemochromatosis is similar to other genetic iron overload disorders such as haemochromatosos associated with mutations in the transferrin receptor two gene, hemojuvelin gene or the hepcidin gene, which supports the role of these gene products in the iron sensing mechanism, that is being unravelled."
Hepcidin
Eventually I will have a whole page devoted to this topic. But for now, I wanted to capture another abstract from the same conference mentioned directly above. The author in this case is Elizabeta Nemeth from UCLA. Again I have copied the full text here in case the link is broken after the conference is completed. Emphasis is added by me.
"The hepatic peptide hormone hepcidin is the principal regulator of iron absorption and tissue iron distribution. Hepcidin circulates in blood plasma and acts at nanomolar concentrations by inducing degradation of its receptor, the cellular iron exporter ferroportin. Ferroportin exports iron into plasma from absorptive enterocytes, from macrophages that recycle the iron of senescent erythrocytes, and from hepatocytes that store iron. Therefore, hepcidin-mediated degradation of ferroportin results in decreased iron absorption in the duodenum, regulating total body iron, as iron losses from the body are normally very small. Hepcidin effect on macrophage ferroportin inhibits the large flux of recycled iron into plasma and decreases plasma iron concentration, as iron is consumed for erythropoiesis and other processes. Hepcidin therefore acts as an endocrine regulator of total body iron stores and plasma iron concentration.
The synthesis of hepcidin is transcriptionally regulated by iron, erythropoiesis and inflammation. Extracellular and intracellular iron concentrations increase hepcidin transcription through a mechanism dependent on the bone morphogenetic protein pathway. Increased iron requirements of erythroid precursors for hemoglobin synthesis cause hepcidin suppression by an unknown pathway. Hepcidin production is also increased by inflammation, primarily through IL6. Dysregulation of these mechanisms leads to aberrant hepcidin production and the development of iron disorders.
Increased hepcidin concentrations in plasma cause or contribute to the pathogenesis of iron-restricted anemias including anemias associated with inflammation (rheumatoid arthritis, inflammatory bowel disease, obesity), chronic kidney disease, some cancers and iron-refractory iron deficiency anemia. Hepcidin deficiency causes iron overload in hereditary hemochromatosis as well as iron-loading anemias such as beta-thalassemia. The hepcidin–ferroportin axis is the principal regulator of extracellular iron homeostasis in health and disease, and is a promising target for the diagnosis and treatment of iron disorders.
Declaration of interest
I am a co-founder and Chief Scientific Officer of Intrinsic LifeSciences, a biotech company developing hepcidin diagnostics. I am also a co-founder of Merganser Biotech, a biotech company developing hepcidin-targeted therapeutics.
Declaration of funding
This work was supported by grants from the National Institute of Health."
"The hepatic peptide hormone hepcidin is the principal regulator of iron absorption and tissue iron distribution. Hepcidin circulates in blood plasma and acts at nanomolar concentrations by inducing degradation of its receptor, the cellular iron exporter ferroportin. Ferroportin exports iron into plasma from absorptive enterocytes, from macrophages that recycle the iron of senescent erythrocytes, and from hepatocytes that store iron. Therefore, hepcidin-mediated degradation of ferroportin results in decreased iron absorption in the duodenum, regulating total body iron, as iron losses from the body are normally very small. Hepcidin effect on macrophage ferroportin inhibits the large flux of recycled iron into plasma and decreases plasma iron concentration, as iron is consumed for erythropoiesis and other processes. Hepcidin therefore acts as an endocrine regulator of total body iron stores and plasma iron concentration.
The synthesis of hepcidin is transcriptionally regulated by iron, erythropoiesis and inflammation. Extracellular and intracellular iron concentrations increase hepcidin transcription through a mechanism dependent on the bone morphogenetic protein pathway. Increased iron requirements of erythroid precursors for hemoglobin synthesis cause hepcidin suppression by an unknown pathway. Hepcidin production is also increased by inflammation, primarily through IL6. Dysregulation of these mechanisms leads to aberrant hepcidin production and the development of iron disorders.
Increased hepcidin concentrations in plasma cause or contribute to the pathogenesis of iron-restricted anemias including anemias associated with inflammation (rheumatoid arthritis, inflammatory bowel disease, obesity), chronic kidney disease, some cancers and iron-refractory iron deficiency anemia. Hepcidin deficiency causes iron overload in hereditary hemochromatosis as well as iron-loading anemias such as beta-thalassemia. The hepcidin–ferroportin axis is the principal regulator of extracellular iron homeostasis in health and disease, and is a promising target for the diagnosis and treatment of iron disorders.
Declaration of interest
I am a co-founder and Chief Scientific Officer of Intrinsic LifeSciences, a biotech company developing hepcidin diagnostics. I am also a co-founder of Merganser Biotech, a biotech company developing hepcidin-targeted therapeutics.
Declaration of funding
This work was supported by grants from the National Institute of Health."
